
¿Por qué la obesidad causa diabetes?
El tejido adiposo constituye la principal reserva de combustible y fuente de energía crítica cuando la comida escasea. El mismo produce y secreta adipoquinas y exosomas involucrados en la regulación de funciones fisiológicas como el apetito, la función reproductiva y la acción de la insulina.
La acumulación excesiva de grasa corporal induce anomalías metabólicas y enfermedades, incluidas la resistencia a la insulina (RI), la dislipidemia aterogénica, la enfermedad del hígado graso no alcohólico, la disfunción de las células beta, la prediabetes y la diabetes mellitus tipo 2 (DM2).
Un progresivo aumento del índice de masa corporal (IMC), se asocia con un mayor riesgo de desarrollar DM2. Sin embargo, la distribución de la grasa modifica el riesgo de la disfunción metabólica inducida por adiposidad.
Aquellas personas con obesidad y con distribución grasa intraabdominal tienen un mayor riesgo de desarrollar DM2 que aquellos con fenotipo graso gluteofemoral.
La DM2 es causada por una resistencia de múltiples órganos a la insulina, sumado a una disminución en la función secretora de insulina de las células beta.
El aumento en la prevalencia de la obesidad es probablemente responsable del creciente aumento en la prevalencia de DM2.
Fisiología celular Beta y cinética de la insulina
Las células beta pancreáticas secretan insulina a la vena porta para entregar al hígado. La concentración de insulina en plasma está determinada por un balance entre la secreción de insulina y la tasa de remoción por el hígado y tejidos extrahepáticos.
En las personas con obesidad, se observa un incremento en la concentración de insulina basal y postprandial, debido al aumento de la secreción pancreática de insulina y la disminución del clearence de insulina portal y periférico. Este aumento en la secreción y concentración de insulina a menudo es capaz de exceder la resistencia a la acción de la insulina, de modo que la concentración de glucosa en sangre y la prueba de tolerancia oral a la glucosa, sean normales.
Sin embargo, la disminución progresiva de la función de las células beta provoca una disminución en el control glucémico, lo que resulta en prediabetes y, en última instancia, DM2.
El incremento en la concentración de ácidos grasos libres plasmáticos (AGL) asociados con obesidad y DM2, puede tener efectos adversos en la célula beta.
Biología del tejido adiposo y resistencia a la insulina (RI)
El tejido adiposo debe tener flexibilidad metabólica, para ser capaz de regular cambios en el balance energético durante la alimentación y el ayuno, y para adaptarse a cambios a largo plazo que causan expansión o reducción del tejido adiposo. El aumento de tejido adiposo producto de un balance energético positivo en forma crónica, causa una acumulación de triglicéridos en los adipocitos, provocando un aumento de su tamaño y remodelación en su estructura. La respuesta adaptativa especifica del tejido adiposo a esta expansión, es un determinante de la salud del tejido adiposo y de su función metabólica, y las diferencias en esta respuesta probablemente contribuyan a la heterogeneidad de la salud metabólica observada en las personas con obesidad.
Los procesos biológicos en el tejido adiposo que contribuyen a la RI incluyen (Figura 1):
- Hipoxia de adipocitos debido al inadecuado suministro de oxígeno y al aumento de la demanda del mismo, que estimula la fibrogénesis del tejido adiposo y la quimiotaxis de macrófagos.
- Mayor número y proporción relativa de células inmunitarias proinflamatorias del tejido adiposo (macrófagos y células T) y la expresión de genes que codifican proteínas proinflamatorias.
- Disminución de la producción y la secreción de adiponectina, hormona sensibilizadora de la insulina.
- Aumento de la actividad lipolítica del tejido adiposo y la liberación de AGL en la circulación.
- Alteraciones metabólicamente nocivas en el contenido de carga de exosomas derivados de macrófagos del tejido adiposo.
Un bajo grado de inflamación crónica, manifestado por el incremento de células inmunes proinflamatorias y la expresión de genes que codifican proteínas proinflamatorias, está presente en las personas con obesidad. Se ha propuesto que la inflamación adiposa es un importante impulsor de la RI en personas con obesidad. Además, un aumento en la grasa visceral es un importante contribuyente a la RI porque genera una mayor liberación de AGL directamente en la circulación portal.
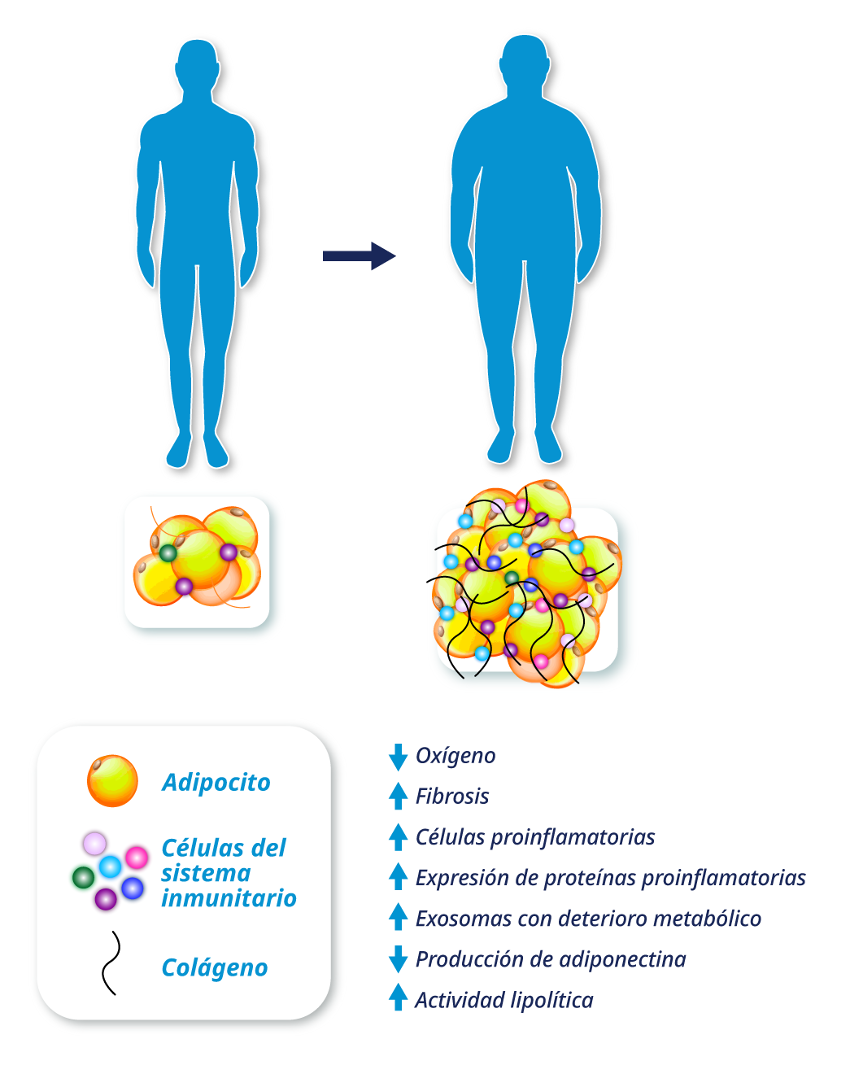
Figura 1: Alteraciones en la biología del tejido adiposo asociadas a disfunción metabólica en personas con obesidad.
Metabolismo hepático de glucosa y lípidos
Durante las condiciones basales, el 80% de la producción de glucosa endógena deriva de la glucogenólisis hepática (producción de glucosa por la descomposición del glucógeno hepático) y gluconeogénesis (glucosa producida a partir de precursores como el lactato, glicerol y aminoácidos) cuya función es mayor en personas con obesidad o DM2.Un aumento en la gluconeogénesis es responsable de la hiperglucemia en ayunas, y la alteración de la supresión de la producción de glucosa endógena. Además, la gluconeogénesis después de la ingestión de comida, contribuye a la hiperglucemia posprandial en personas con prediabetes y DM2. Las personas con obesidad suelen tener un deterioro en la capacidad de la insulina para suprimir la producción hepática de glucosa, pero, a menudo, tienen tasas normales de producción de glucosa hepática basal y posprandial debido al aumento de la secreción de insulina.
La RI en el tejido adiposo tiene efectos indirectos sobre el metabolismo hepático de la glucosa porque la supresión alterada de la lipólisis del tejido adiposo aumenta la liberación de AGL que son entregados al hígado, lo que aumenta la gluconeogénesis hepática.
Metabolismo de la glucosa en el sistema musculoesquelético
Durante las condiciones basales, los AGL del plasma sirven como combustible muscular. Después de la ingesta de glucosa o comida mixta, el aumento de la insulina plasmática suprime la lipólisis de triglicéridos del tejido adiposo, disminuye los AGL y estimula la captación de glucosa muscular, que provoca un cambio en el combustible muscular de los ácidos grasos a la glucosa. La captación de glucosa muscular estimulada por insulina implica la unión de la misma a sus receptores en los miocitos, lo que inicia una cascada de eventos de señalización intracelular que resultan en la ubicación del transportador de glucosa 4 en membrana plasmática, necesario para el transporte de glucosa al interior de la célula. Al entrar, la glucosa del miocito se fosforila y puede oxidarse para obtener combustible a través de la glucólisis o almacenarse como glucógeno. Las personas con obesidad y las personas con DM2 tienen un deterioro tanto en la oxidación de glucosa muscular como en la síntesis de glucógeno causada por una regulación en baja del número y función de los receptores de insulina y múltiples defectos en la señalización posterior al receptor.
Efecto de la pérdida de peso
La pérdida de peso puede tener profundos efectos terapéuticos en la función metabólica, DM2 y las enfermedades asociadas. Una disminución moderada del 5% al 10% del peso corporal mejora el control glucémico, las concentraciones plasmáticas de triglicéridos, colesterol HDL, y la presión arterial. La pérdida de peso necesaria para lograr máximos beneficios metabólicos entre diferentes sistemas diferirá de persona a persona según la severidad y duración de la anormalidad metabólica.
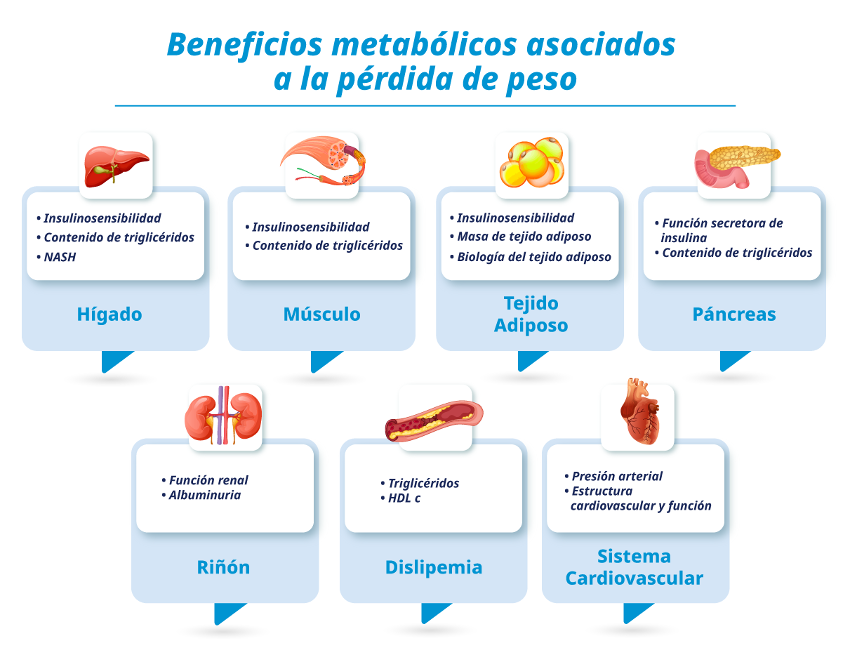
Figura 1: La pérdida de peso y sus beneficios metabólicos.
Conclusión:
La obesidad, particularmente cuando se asocia con aumento de grasa intraabdominal e intrahepática, es un factor de riesgo para el desarrollo de prediabetes y DM2 porque causa tanto RI como disfunción de las células beta. En consecuencia, el aumento en la prevalencia de la obesidad ha llevado al concomitante aumento de la prevalencia de DM2. La disminución de la masa grasa corporal puede mejorar la disfunción metabólica inducida por la obesidad e incluso lograr remisión de la DM2.Bibliografía:
Cita: Cell Metabolism 34, January 4, 2022
Klein S, Gastaldelli A, Yki-Järvinen H, Scherer PE. Why does obesity cause diabetes?
Cell Metab. 2022 Jan 4;34(1):11-20. doi: 10.1016/j.cmet.2021.12.012.
Codigo de Aprobación: AR23OB00004
Disclaimer
Material desarrollado con fines de educación Médica continua.
Material exclusivo para profesionales de la salud.
La información aquí presentada es opinión del ponente.
NetMD Education © 2023
Entradas recientes
