Esperanza para los pacientes con déficit de esfingomielinasa ácida, una enfermedad ultra-rara, grave y debilitante
- ComiteNetMD
- 28 de octubre de 2024
- Endocrinología y Diabetes, Medicina General e Interna, Pediatria
- 0 Comments
Se estima que en el mundo hay alrededor de 1.000 personas afectadas por el déficit de esfingomielinasa ácida (ASMD), de las cuales varias decenas son españolas. Hasta hace pocos meses, los pacientes de nuestro país no disponían de opciones terapéuticas específicas para la enfermedad.
Coincidiendo con el Día Mundial de Concienciación del ASMD (19 de octubre), la comunidad española en torno a esta enfermedad ultra-rara celebra el último avance en su abordaje: un nuevo paradigma en el tratamiento del déficit de esfingomielinasa ácida (nombre completo de este trastorno minoritario), tras años de investigaciones y reivindicaciones tanto por parte de la comunidad científico-médica como de los pacientes y su entorno. Hasta hace pocos meses, las varias decenas de afectados que se estima que hay en España sólo contaban con opciones terapéuticas limitadas a cuidados sintomáticos y de apoyo.
El ASMD es una enfermedad genética y progresiva que afecta tanto a bebés y niños como a adultos, comprometiendo severamente su calidad de vida debido a su diversidad de síntomas y su afectación multiorgánica. Unas características que pueden impactar de forma considerable en la calidad de vida de quienes viven con esta patología poco frecuente, limitando su capacidad para realizar actividades cotidianas y, en los casos más graves, llevándolos a una situación de discapacidad o incluso provocándoles la muerte prematura.
Dra. Montserrat Morales
Coordinadora de la Unidad de Adultos de Enfermedades Raras y Errores Congénitos del Metabolismo del Hospital Universitario 12 de Octubre (Madrid)
“Estamos ante un hito significativo para los pacientes con ASMD, quienes hasta ahora carecían de opciones terapéuticas específicas, limitándose a tratamientos sintomáticos. Este avance en el abordaje de la enfermedad mejora sustancialmente su calidad de vida al reducir síntomas. Asimismo, podría transformar el pronóstico a largo plazo de los pacientes adultos con ASMD, mejorando aspectos críticos e incapacitantes, que tradicionalmente acortaban su esperanza de vida. Además, se anticipa que estos avances terapéuticos también ofrecerán una nueva esperanza para quienes viven con esta enfermedad devastadora”.
Existen varios tipos de déficit de esfingomielinasa ácida: el ASMD tipo A, que suele ser más grave y afectar a bebés; y el ASMD tipo B, que suele manifestarse en la infancia tardía o la edad adulta. También hay el ASMD tipo A/B, que es una fórmula intermedia.
Dr. Antonio González-Meneses López
Pediatra especialista en enfermedades raras de causa genética en el Hospital Universitario Virgen del Rocío (Sevilla)
“El nuevo paradigma de tratamiento va a tener un impacto importantísimo en los niños afectados por ASMD tipo B, evitando su progresión, mejorando sus síntomas y, lo más importante, reduciendo la mortalidad precoz. Este avance ofrece una esperanza renovada para los pacientes y sus familias”.
Compromiso con la I+D
Como parte de su propósito de perseguir el poder de la ciencia para mejorar la salud de las personas, Sanofi apuesta por la investigación en el campo de las enfermedades raras (EERR) con especial atención en las de depósito lisosomal y las hematológicas.
Raquel Tapia
Directora general de Sanofi Iberia
“Nuestro compromiso con la I+D para mejorar la vida de las personas con enfermedades raras es real. A modo de ejemplo, en la actualidad el 14% de todos los proyectos en investigación que la Compañía tiene en marcha en el mundo son para enfermedades minoritarias, lo que nos mantiene como referentes globales en este campo”.
La Compañía fue pionera mundial y en España en desarrollar una solución terapéutica para una enfermedad rara de depósito lisosomal (en concreto para la enfermedad de Gaucher), convirtiéndose en un referente en el área de las patologías minoritarias. Fue además la primera terapia de reemplazo enzimático, un tipo de tratamiento específico para enfermedades genéticas que, como el ASMD, se caracterizan por la carencia de una enzima: ésta se administra artificialmente para corregir su déficit y restaurar la función normal del organismo.
Sobre el ASMD4
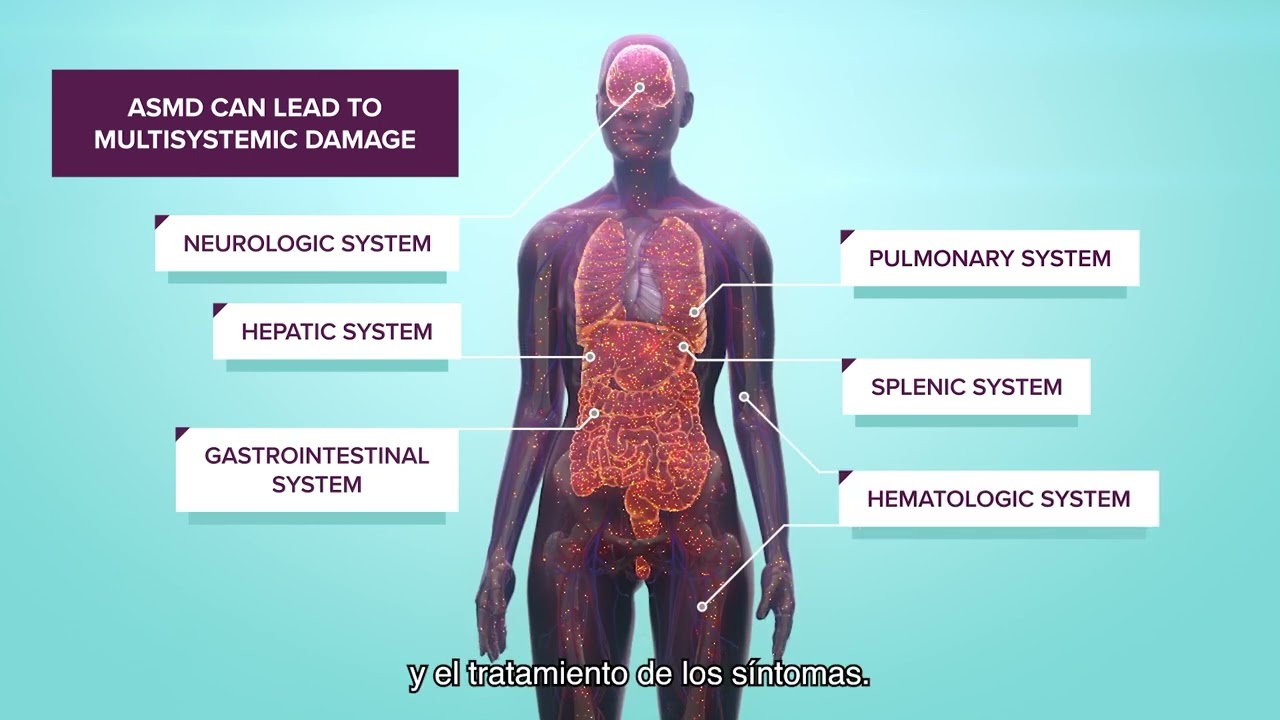
Históricamente conocida como enfermedad de Niemann-Pick tipos A, A/B y B, el ASMD (acrónimo de Acid Sphingomyelinase Decifiency) o déficit de esfingomielinasa ácida es una enfermedad ultra-rara de origen genético, progresiva y potencialmente fatal. Forma parte del grupo de enfermedades de depósito lisosomal -o lisosomales- y se caracteriza por la falta de la enzima esfingomielinasa ácida (ASM), la cual permite la descomposición de la esfingomielina lipídica. Una cantidad insuficiente de la enzima ASM implica que la esfingomielina está mal metabolizada, lo que podría ocasionar una acumulación de por vida en múltiples órganos (pulmón, hígado o bazo, entre otros), causando su mal funcionamiento.
Estas complicaciones pueden afectar significativamente la calidad de vida de los pacientes, limitando su capacidad para realizar actividades cotidianas y, en los casos más graves, llevándolos a una situación de discapacidad o, en última instancia, provocándoles la muerte prematura1-3.
El ASMD engloba dos tipos que pueden suponer extremos opuestos: ASMD tipo A y ASMD tipo B. La primera forma es más grave porque implica afectación neurológica progresiva, además de afectación multiorgánica, y suele manifestarse los primeros meses de vida, condicionando una muerte temprana. La segunda tiene una edad de diagnóstico variable, aunque suele comenzar en la infancia tardía o la edad adulta. Por otra parte, el ASMD tipo A/B es una forma intermedia que incluye diversos grados de afectación del sistema nervioso central (SNC).
El déficit de esfingomielinasa ácida tiene una incidencia aproximada de un caso entre 250.000 nacidos vivos. Su prevalencia en España se está estudiando, aunque en este momento hay diagnosticados varias decenas de pacientes.