Osteogénesis Imperfecta.
- netmd
- 19 de julio de 2017
- Reumatología
- 0 Comments
Fundamento: La Osteogénesis Imperfecta (OI) constituye la forma más frecuente de enfermedad esquelética de causa genética. Es un desorden hereditario del tejido conectivo caracterizado por fragilidad y disminución de la masa ósea y fracturas múltiples. Esta patología puede producir deformaciones y hasta sordera en algunos casos que en ocasiones limitan la vida social de los pacientes.
Autores:
Laura Beatriz Antúnez Torres. Estudiante de 4to año de la carrera de medicina. Instituto Superior de Ciencias Médicas Martha Abreu de Villa Clara. Facultad de Ciencias Médicas: Dr. Faustino Pérez Hernández. Sancti Spíritus. Cuba.
Dr. Jorge Enrique Pérez Díaz. Especialista de Primer grado en Medicina General Integral. Máster en Emergencias Médicas. Profesor Asistente. Instituto Superior de Ciencias Médicas Martha Abreu de Villa Clara. Facultad de Ciencias Médicas: Dr. Faustino Pérez Hernández. Policlínico Juan Latinan Aguilar. Banao. Sancti Spíritus. Cuba..
Dra. Yamilse María Torres León. Especialista de Primer grado en Medicina General Integral. Profesor Asistente. Máster en Emergencias Médicas. Instituto Superior de Ciencias Médicas Martha Abreu de Villa Clara. Facultad de Ciencias Médicas: Dr. Faustino Pérez Hernández. Policlínico Juan Latinan Aguilar. Banao. Sancti Spíritus. Cuba
Resumen
El objetivo de este artículo es revisar el tema a raíz de la presentación de un caso.
Presentación de caso: Se describe el caso de un escolar de 11 años de edad, que ha sufrido 4 fracturas en diversos huesos sin recibir traumas severos que pudieran ocasionarlas.
Conclusiones: La Osteogénesis Imperfecta (OI) es una patología difícil de diagnosticar si no se piensa en ella, por lo que su diagnóstico es tardío en ocasiones. Es de vital importancia el manejo médico y familiar correcto de la misma.
Palabras Claves: osteogénesis imperfecta, fracturas, densidad ósea.
Introducción.
La Osteogénesis Imperfecta (OI) es una patología hereditaria no tan infrecuente, existiendo actualmente en países como Estados Unidos un estimado de 20.000 a 50.000 pacientes con éste diagnóstico. 1 Tiene un rango de severidad variable, desde formas letales en el período neonatal a fracturas ocasionales. No existe cura para ella, aunque se utilizan actualmente algunos medicamentos con buenos resultados. Por lo tanto el tratamiento fundamental es la prevención y control de los síntomas y desarrollar huesos más fuertes, para lo cual se orientan ejercicios como la natación.
Se describe el caso de un escolar de 11 años de edad al que se le diagnostica la enfermedad.
Presentación de caso
Escolar MSD, masculino, de 11 años de edad, procedente de área rural, que es ingresado en el servicio de ortopedia por fractura del fémur izquierdo luego de recibir una contusión directa en dicha área. Antecedentes familiares: paternos hipertensión arterial, maternos no refiere. Interrogatorio positivo: fractura de clavícula derecha durante el parto, fractura de radio derecho luego de torsión del antebrazo hace 11 meses, fractura de ulna izquierda por caída desde sus pies hace 8 meses y fractura de falange de la mano derecha hace 4 meses por golpear una pelota de beisbol. Examen Físico positivo en estos momentos: inflamación y dolor a nivel del muslo izquierdo, se constata hipotonía muscular generalizada, laxitud articular, escoliosis y coloración gris de la esclera con ligera protrusión de los ojos.
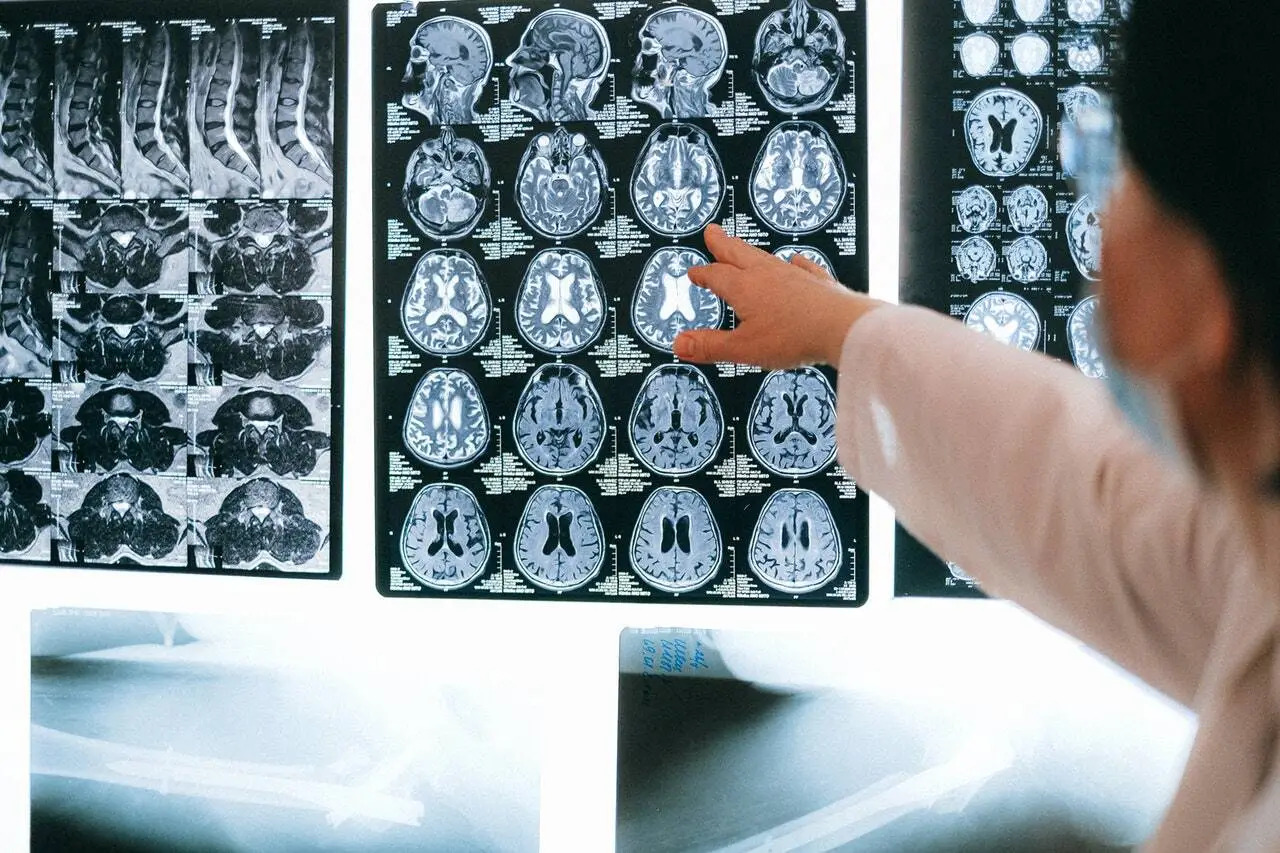
Complementarios: Hemograma normal, creatinina 70 g/l, Urea 6.9 mg/dl, calcio 10 mg/dl, fósforo 3 mg/dl, magnesio 2.6 mEq/L, fosfatasa alcalina 781 U/l, proteínas totales 7.9 mg/dl, albúmina 5.5 g/dl, examen general de orina normal, creatinina en orina 330, densitometría ósea que reporta T-score de – 2,5 compatible con osteoporosis. Radiografía de huesos de pierna y muslo muestran cambios en su morfología, disminución en el grosor de las corticales, bandas transversales en los huesos tubulares y desmineralización generalizada. Diagnóstico: cambios óseos sugerentes a síndrome de Osteogénesis Imperfecta.
Se somete a cirugia y se toma muestra para estudio anatomopatológico el cual corrobora el diagnostico. Se informa a la familia sobre los cuidados a tener con el paciente y se da asesoramiento genético. Se comienza de inmediato el tratamiento medicamentoso del mismo.
Discusión
La osteogénesis imperfecta es una enfermedad caracterizada por fractura de los huesos fácilmente. El predominio de Osteogénesis Imperfecta (OI) es aproximadamente 16 casos por millón de habitantes. La herencia es generalmente autosómica dominante, pero son comunes mutaciones nuevas y también se presentan casos de herencia autosómico recesiva. 2
Sus síntomas son variados y según ellos se clasifica la enfermedad. La clasificación más ampliamente utilizada es la de Sillence que incluye cuatro tipos de Osteogénesis Imperfecta (OI) basados en sus características clínicas, radiológicas y su severidad 3. Sin embargo, en la actualidad han sido delineados otros tres grupos de pacientes que presentan un diagnóstico clínico de Osteogénesis Imperfecta (OI), pero se observa en ellos rasgos claramente diferentes 4.
El tipo I Osteogénesis Imperfecta (OI) es en gran medida el subtipo clínico más común, excepto en África meridional, donde es más común el tipo III, es precisamente el caso que nos ocupa según la clínica que presenta. La evaluación de la signología que presentan los pacientes, permite reconocer signos de mayor y menor criterio para establecer el diagnóstico 5.. Entre los primeros tenemos las escleras azules, la dentinogénesis imperfecta y la osteoporosis, constituyendo la triada clásica 6.
Como signos de menor criterio se consideran la sordera, laxitud ligamentaria, escoliosis, facilidad para formar equimosis, sudoración excesiva y estreñimiento. La osteoporosis es el signo cardinal, siempre presente, que se acompaña de deformaciones de los huesos largos a consecuencia de las fracturas, y de xifoescoliosis en la columna por fracturas vertebrales. Las escleróticas azules se producen por adelgazamiento del colágeno. La dentinogénesis imperfecta presente en la Osteogénesis Imperfecta (OI) representa una anormalidad de la dentina; los dientes son cortos, decolorados (usualmente de color marrón). La sordera que aparece al final de la segunda década de la vida, se atribuye a cambios degenerativos en el oído medio a nivel de los huesecillos 7.
El diagnostico es considerado en diferentes momentos de la vida: durante el desarrollo fetal, al nacimiento, en la niñez y menos frecuentemente en la edad adulta y es fundamentalmente clínico 3 aunque la radiografía contribuye al mismo 2, Las imágenes características pasan por mostrar metáfisis coraliformes quísticas por defectos de remodelación cuando el niño no ha caminado, imágenes similares a palomitas de maíz en metáfisis y epífisis cerca de las laminas de crecimiento, zonas radiolúcidas redondeadas con bordes escleróticos. En las formas menos graves: osteoporosis, corticales finas y en el conducto medular las trabéculas muy finas con imagen de plumas, disminución de la remodelación ósea en metáfisis dando imagen de trompeta.
Se observa una desmineralización generalizada, los huesos presentan una cortical muy delgada y una esponjosa pobremente trabeculada, existe la evidencia de fracturas recientes o antiguas en diversos estados de consolidación. En la variedad congénita y de carácter recesivo la cortical de las diáfisis de los huesos largos aparecen ensanchadas de forma excepcional. Igualmente es evidente la deformación de las metáfisis muy ensanchadas. El cráneo presenta forma de hongo, bóveda muy delgada, osificación escasa y tardía con severo retardo en su osificación y un mosaico típico de huesos wormianos 8.
Tambien la densitometría ósea es una ayuda de diagnóstico cuando falta la clínica y la radiología. Niños sanos que tienen más de 3 fracturas de baja energía en un año deben ser sometidos a densitometría ósea como parte de un rastreo de Osteogénesis Imperfecta (OI). Los niños con Osteogénesis Imperfecta (OI) tienen a nivel del cuello femoral disminuida la BMD (Bone Mineral Density) en comparación con niños sanos de la misma edad y peso. Se ha postulado que los pacientes con OI tienen deficiencias en la mineralización, secundariamente a las anomalías de la síntesis del tipo I del colágeno. La densitometría ósea ha demostrado ser una herramienta importante para el diagnóstico y monitoreo de la densidad ósea, pues tiene un bajo costo relativo, una baja exposición a la radiación y una técnica precisa que permite el seguimiento de los pacientes. Los estudios clínicos de la osteoporosis en adultos han demostrado que los valores bajos de BMD están asociados con el riesgo de fracturas. 9
En cuanto al tratamiento históricamente no existía otro que el manejo del dolor y las deformaciones. Actualmente tenemos dos opciones farmacológicas para incrementar la masa ósea. Una es incrementando la actividad osteoblástica con análogos de la hormona paratiroidea y la otra disminuyendo la actividad osteoclástica con bifosfonatos, la primera opción está contraindicada en niños por el peligro de producir osteosarcoma a nivel de los cartílagos de crecimiento en plena actividad 10.
Se ensayaron múltiples drogas y diferentes tipos de tratamiento para evitar el progreso de la enfermedad, sin resultado satisfactorio hasta la aparición de los bifosfonatos, que son los agentes antiresortivos clínicos más importantes para tratar enfermedades caracterizadas por aumento del ciclo de reabsorción mediado por los osteoclastos. Las enfermedades más comunes que tienen esta característica son la Osteoporosis, la Enfermedad de Paget y los estadíos de enfermedades metastásicas por cáncer. El tratamiento de enfermedades pediátricas como Osteogénesis Imperfecta (OI) y displasia fibrosa, al momento tienen una aceptable seguridad, aunque falta un seguimiento a largo plazo 11. La actual literatura médica está repleta de series de tratamiento satisfactorio con bifosfonatos, de tal forma que son ampliamente usados para el tratamiento de Osteogénesis Imperfecta (OI) en niños, adolescentes y adultos 12.
También está indicada la administración de sulfato y oxido de magnesio, vitaminas C y D, fluoruros y más recientemente la calcitonina 13. Con el objeto de ayudar a la mejor consistencia del esqueleto se aconseja estimular precozmente la estación de pie y el comienzo de la marcha, lo cual usualmente requiere uso de aparatos ortopédicos. Se han usado soportes como pantalones al vacío los cuales además previenen la aparición de fracturas mejorando en lo posible la densidad del hueso.
Desde el punto de vista ortopédico el tratamiento se dirige al cuidado de las fracturas. Si bien es cierto que las fracturas en la Osteogénesis Imperfecta (OI) consolidan en el tiempo normal, es un hecho reconocido que en la zona correspondiente persiste aun más fragilidad que en el resto del hueso primitivo, las deformidades deben ser evitadas en lo posible pero una vez establecidas hay que corregirlas mediante procedimientos quirúrgicos.
Conclusiones
La Osteogénesis Imperfecta (OI) es una patología difícil de diagnosticar si no se piensa en ella, por lo que su diagnóstico es tardío en ocasiones. Es de vital importancia el manejo médico y familiar correcto de la misma.
Bibliografía
- James, William; Berger, Timothy; Elston, Dirk. Andrews’ Diseases of the Skin: Clinical Dermatology 2005;(10th Ed.). Page 517. ISBN 0-7216-2921-0.
- Charnas, L. R.; Marini, J. C. “Communicating hydrocephalus, basilar invagination, and other neurologic features in osteogenesis imperfecta”. Neurology 1999;43 (12): 2603–2608. ISSN0028-3878. PMID 8255464.
- “Pain Management – Osteogenesis Imperfecta Foundation | OIF.org”. www.oif.org. Retrieved 2016-01-06.
- Rauch F, Glorieux FH. “Osteogenesis imperfecta” 2013; Lancet 363 (9418): 1377–85.
- Steiner RD, Pepin MG, Byers PH, Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP. “Osteogenesis Imperfecta”. PMID20301472. Retrieved 26 March 2012.
- Shapiro JR, Lietman C, Grover M, Lu JT, Nagamani SC, Dawson BC, Baldridge DM, Bainbridge MN, Cohn DH, Blazo M, Roberts TT, Brennen FS, Wu Y, Gibbs RA, Melvin P, Campeau PM, Lee BH. “Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation”. Bone Miner 2013; Res. 28 (7): 1523–30.
- Fuller E, Lin V, Bell M, Bharatha A, Yeung R, Aviv RI, Symons SP. “Case of the month #171: osteogenesis imperfecta of the temporal bone”. Can AssocRadiol J 622015; (4): 296–8.
- Chen, Harold. Atlas of genetic diagnosis and counseling. Totowa, NJ: Humana Press.2005;ISBN1-58829-681-4.
- Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F, Bishop NJ. “Type V osteogenesis imperfecta: a new form of brittle bone disease”. Bone Miner2005; Res. 15 (9): 1650–8.
- “Recessive Form of OI Discovered by Foundation-funded Researcher” (PDF).
- Genetics Home Reference: Genetic Conditions >Osteogenesisimperfecta (Reviewed November 2007)
- Volodarsky M, Markus B, Cohen I, Staretz-Chacham O, Flusser H, Landau D, Shelef I, Langer Y, Birk OS. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfect.2013:
- Gautieri A, Uzel S, VesentiniS, Redaelli A, Buehler MJ. “Molecular and mesoscale disease mechanisms of Osteogenesis Imperfecta”. Biophysical Journal 2009; (3): 857–865.