Histiocitosis de células de Langerhans con afectación hipofisaria, seguimiento endocrinológico en paciente adulto: A propósito de un caso
- ComiteNetMD
- 20 de noviembre de 2024
- Endocrinología y Diabetes
- 0 Comments
Resumen:
La histiocitosis de células de Langerhans (HCL) es una rara afección histiocítica que se distingue por el crecimiento excesivo de células del sistema fagocítico mononuclear (macrófagos, células dendríticas, monocitos) en diversos órganos y sistemas. La afectación de la glándula hipófiisis se reporta en 5-50% de los pacientes con HCL, lo más frecuente la deficiencia de arginina vasopresina en 50% de los casos. Una minoría también presenta disfunción de hormonas de la adenohipófisis. Presentamos el caso de un paciente masculino de 33 años, diagnosticado de HCL en contexto de infecciones óticas a repetición, con tratamiento quimioterápico hasta en 3 ocasiones por recidiva tumoral. En el seguimiento se evidencia deficiencia de arginina-vasopresina. Asimismo, se diagnostica panhipopituitarismo. Nos enfocaremos en la definición, fisiopatología, epidemiología, y cómo realizar el seguimiento endocrinológico en pacientes adultos con HCL. La afectación de la hipófisis puede resultar un desafío diagnóstico. Es fundamental reconocer esta manifestación clínica para un diagnóstico y tratamiento adecuado, ya que sin tratamiento el paciente puede enfrentar un riesgo vital.
Introducción
La histiocitosis de células de Langerhans (HCL), asi llamada por la infiltración tisular de células con estructura y marcadores específicos de las células de Langerhans (CD1a+/CD207+), presentes en las lesiones entre un 1% a > 70% (promedio de 8%)1. En 1953, fue agrupada bajo el término histiocitosis X debido al origen incierto de las células. En los años 80, gracias a la microscopía electrónica, se pudo diferenciar los tipos de histiocitosis, lo que llevó a la definición de HCL por parte de la sociedad de histiocitosis2.
Se caracteriza por activación constitutiva de la vía MAPK, lo que produce una multiplicación clonal de precursores mieloides que se diferencian en CD1a+/CD207+ en los tejidos afectados. Estas células tienen una baja tasa de proliferacion pero son menos sensibles a la apoptosis, lo que resulta en lesiones persistentes. Ocurre de manera esporádica3.
La histiocitosis de células de Langerhans (HCL) puede presentarse de varias maneras: unifocalmente, con una lesión única en un solo órgano; multifocalmente en un solo sistema, con dos o más lesiones que afectan un solo órgano; y de forma multisistémica, con dos o más órganos comprometidos4. Se estima una prevalencia entre 1 y 1,5 casos por millón de habitantes/año en población pediátrica2,5. La epidemiología es desconocida en adultos.
El diagnóstico se realiza mediante la histopatología del tejido afecto, junto con inmunohistoquímica que evidencie positividad del marcador CD1a2. Las pautas actuales proponen que tanto para la estadificación inicial como el seguimiento se realicen con tomografía por emisión de positrones con flúor‐18‐fluorodeoxiglucosa (18-PET- FDG)4,5.
Esta patología puede comprometer diversos sistemas, que incluyen el sistema esquelético, pulmonar, timo, sistema hepatobiliar, sistema digestivo, sistema nervioso central (SNC), los tejidos blandos de la cabeza y el cuello, glándulas salivales y raramente tiroides6. Las manifestaciones cutáneas y esqueléticas son las más comunes en pacientes adultos, es especial de cráneo y vertebras.
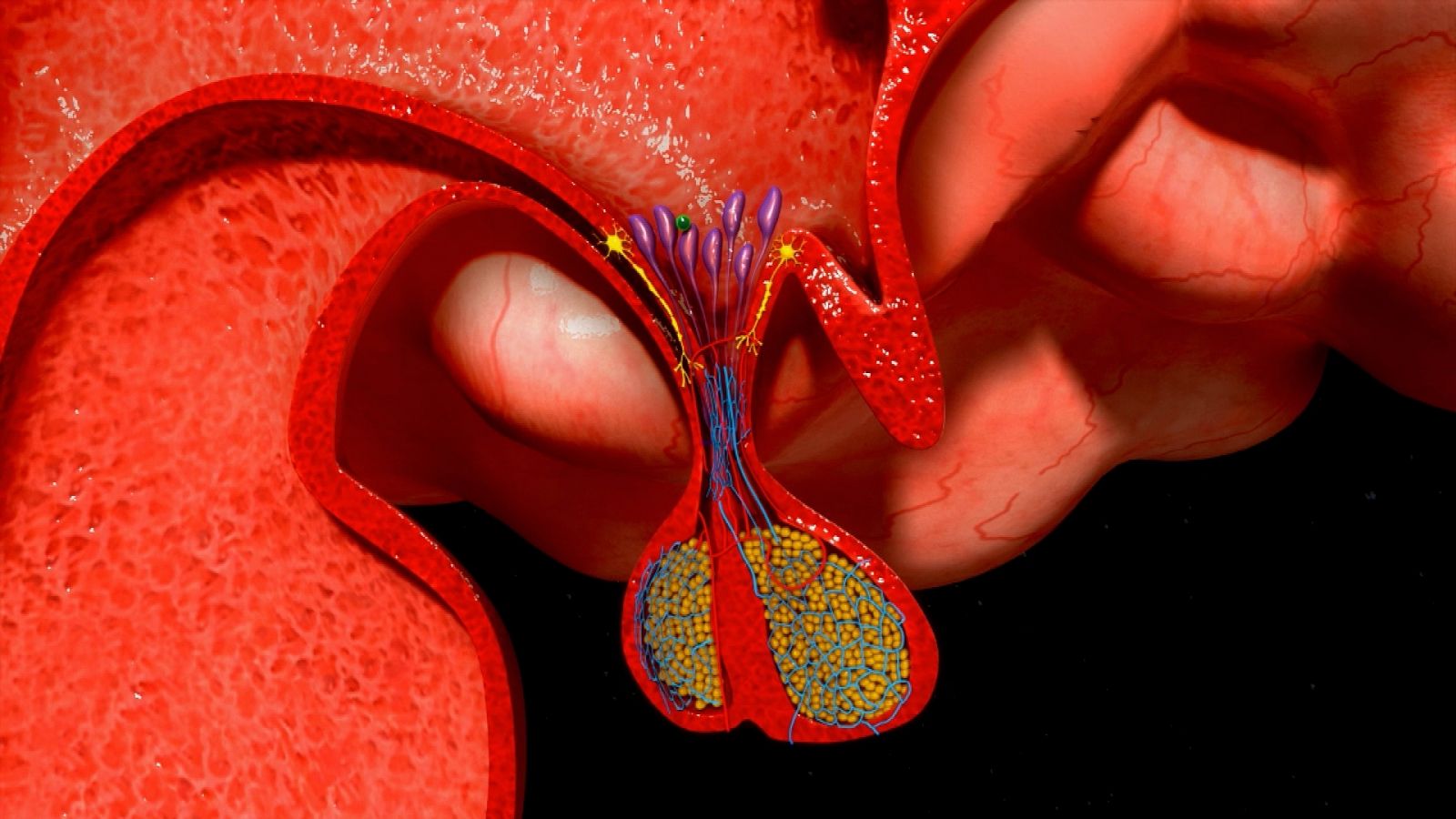
En lo endocrinológico, la afectación de la glándula hipófiisis se reporta en 5-50% de los pacientes con HCL, lo mas frecuente la deficiencia de arginina vasopresina en 50% de los casos7. Una minoría (5-20%) también presenta disfunción de hormonas de la adenohipófisis4,8,9. En la resonancia magnética nuclear (RMN) se puede encontrar a nivel de sistema nervioso central las siguientes manifestaciones: pérdida del punto brillante de la glándula pituitaria, engrosamiento del tallo pituitario >3 mm con realce, lesión masiva con isointensidad y realce homogéneo bajo imágenes potenciadas en T16.
Reportamos el caso de un paciente con HCL diagnosticado en la pubertad que en la evolución de la enfermedad agrega múltiples déficits hormonales. Representa un desafío dada la poca frecuencia de la patología.
Bryan Acosta1, Florencia Dorfman1, María. M. Piñeyro1.
1 Clínica de Endocrinología y Metabolismo, Hospital de Clínicas, Facultad de Medicina, Universidad de la República, Montevideo, Uruguay. Departamento de Nutrición. Facultad de Medicina Norte. Universidad de Chile. Santiago, Chile.
Para descargar la investigación completa haga clik a continuación:
https://www.revistasoched.cl/4_2024/04.html